Background
Structure
Protein kinases (PKs) are enzymes that transfer phosphoryl group from an ATP molecule to Ser, Thr or Tyr residue of the substrate protein. The human genome consists 484 PK genes (497 domains) that are divided broadly into nine families based on their sequences namely, AGC, CAMK, CK1, CMGC, NEK, RGC, STE, TKL, TYR and OTHER (unclassified). They share a conserved structural fold consisting of two lobes: an N-terminal lobe, formed by five stranded β-sheet with an α-helix called the C-helix, and a C-terminal lobe comprising six α-helices. The two lobes are connected by a flexible region in the middle which forms the ATP binding active site of the protein.
Activation loop
The activation loop is typically 20-30 residues in length and is the most critical secondary structural element of the active site of PKs. It is in completely extended conformation in the catalytically active state of the enzyme facilitating the binding of ATP molecule and the substrate. However, it folds on the surface of the protein in different kinds of inactive states. The activation loop begins with a conserved sequence motif called DFGmotif (Asp, Phe, and Gly residues). These residues are observed to be in a unique orientation when the loop is extended (active state) but display remarkable flexibility in folded (inactive) loop conformations.
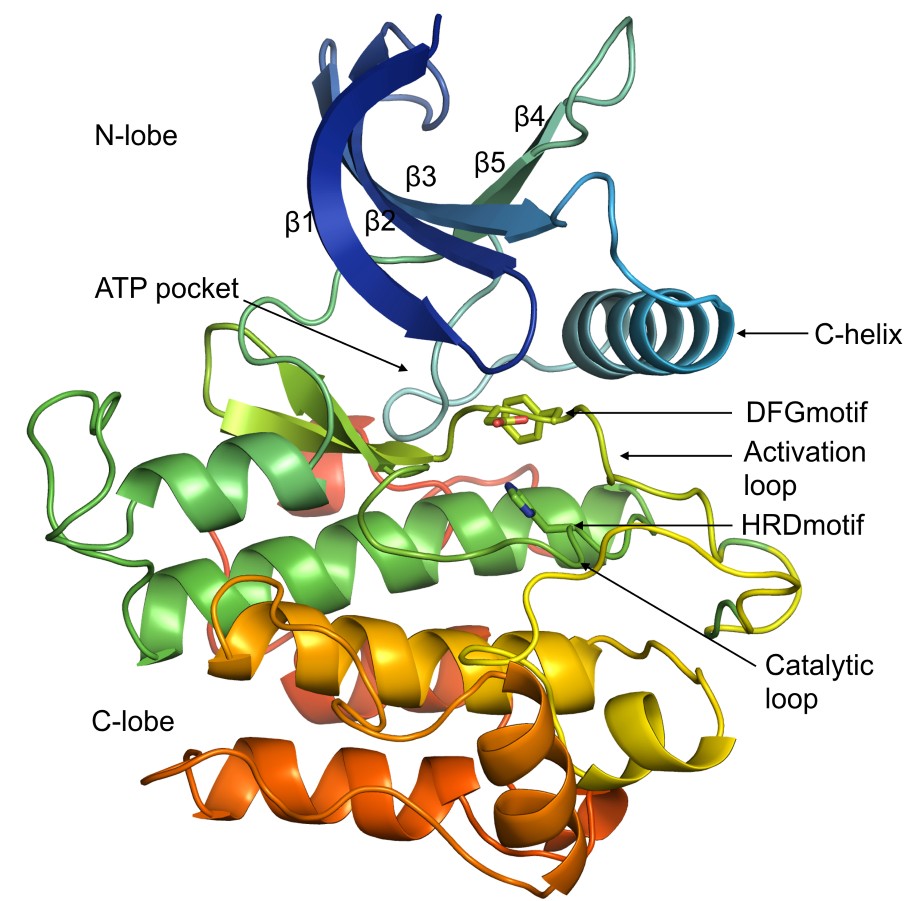
Typical structure of protein kinase
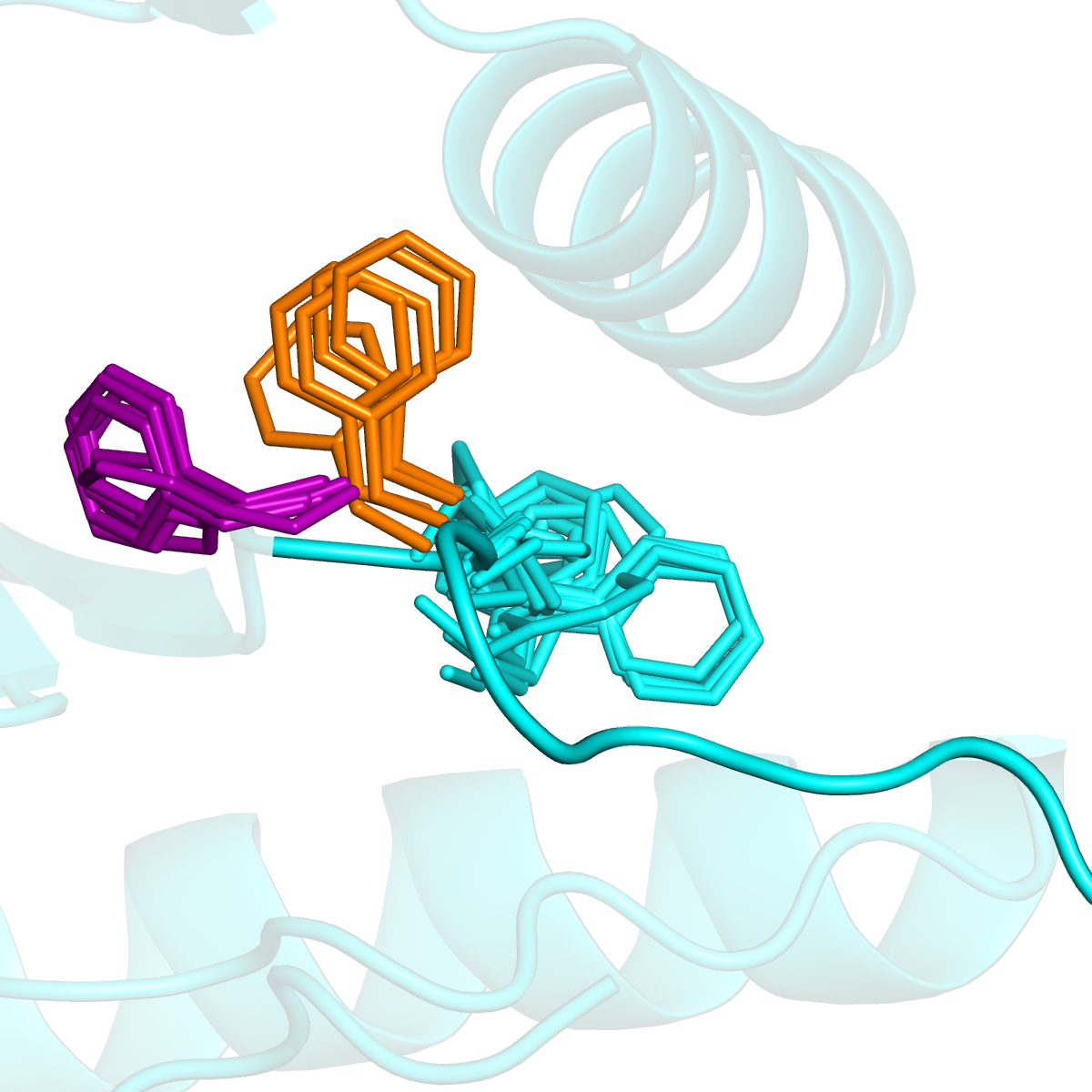
Multiple orientations of DFG-Phe in EGFR
Classification and nomenclature for active and inactive protein kinase structures
We have clustered all the conformations of PK activation loop in two steps:
Spatial groups
We have determined the location of DFG-Phe ring in the binding pocket based on its distance from two
conserved residues,
- D1 = dist(αC-Glu(+4)-Cα, DFG-Phe-Cζ) - distance between the Cα atom of the fourth residue after the conserved Glu residue in the C-helix (ExxxX) and the outermost atom of the DFG-Phe ring (Cζ). This distance serves to distinguish DFGin structures, where the Phe ring is adjacent to or under the C-helix, from DFGout structures, where the ring has moved a substantial distance laterally from the C-helix.
- D2= dist(β3-Lys-Cα, DFG-Phe-Cζ) - distance between the Cα atom of the conserved Lys residue from the β3 strand to the Cζ atom of the DFG-Phe side chain. It captures the closeness of DFG-Phe to the N-lobe β-sheet strands, thus giving an estimate of the upward position of the Phe ring, distinguishing DFGin conformations from structures where the Phe ring is in an intermediate position between DFGin and DFGout.
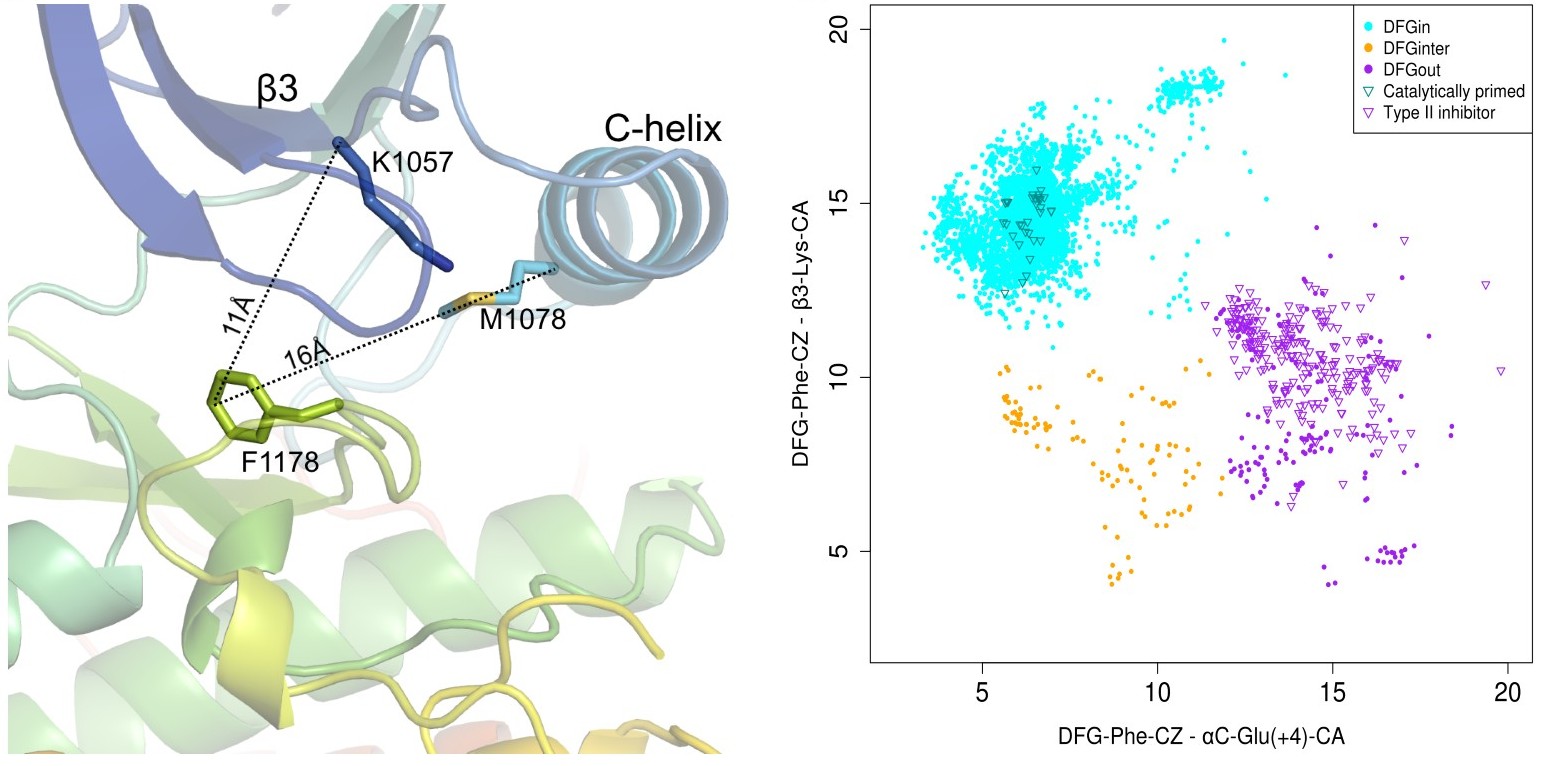
Based on the spatial location of DFG-Phe ring in the binding pocket we have classified kinase structures into three broad groups:
- DFGin: This is the largest group representing the DFG motif orientations where DFG-Phe is packed against or under the C-helix. It consists of many related conformations with the typical DFGin active orientation forming the largest subset of this group.
- DFGout: This is the second largest group representing the structures where DFG-Phe is moved into the ATP binding pocket. The structures with a Type 2 inhibitor bound form a subset of this group.
- DFGinter - (DFGintermediate): This is the smallest group in which the DFG-Phe side chain is out of the C-helix pocket but has not moved completely to a DFGout conformation. In most of these cases DFG-Phe is pointing upwards towards the β-sheets dividing the active site into two halves.
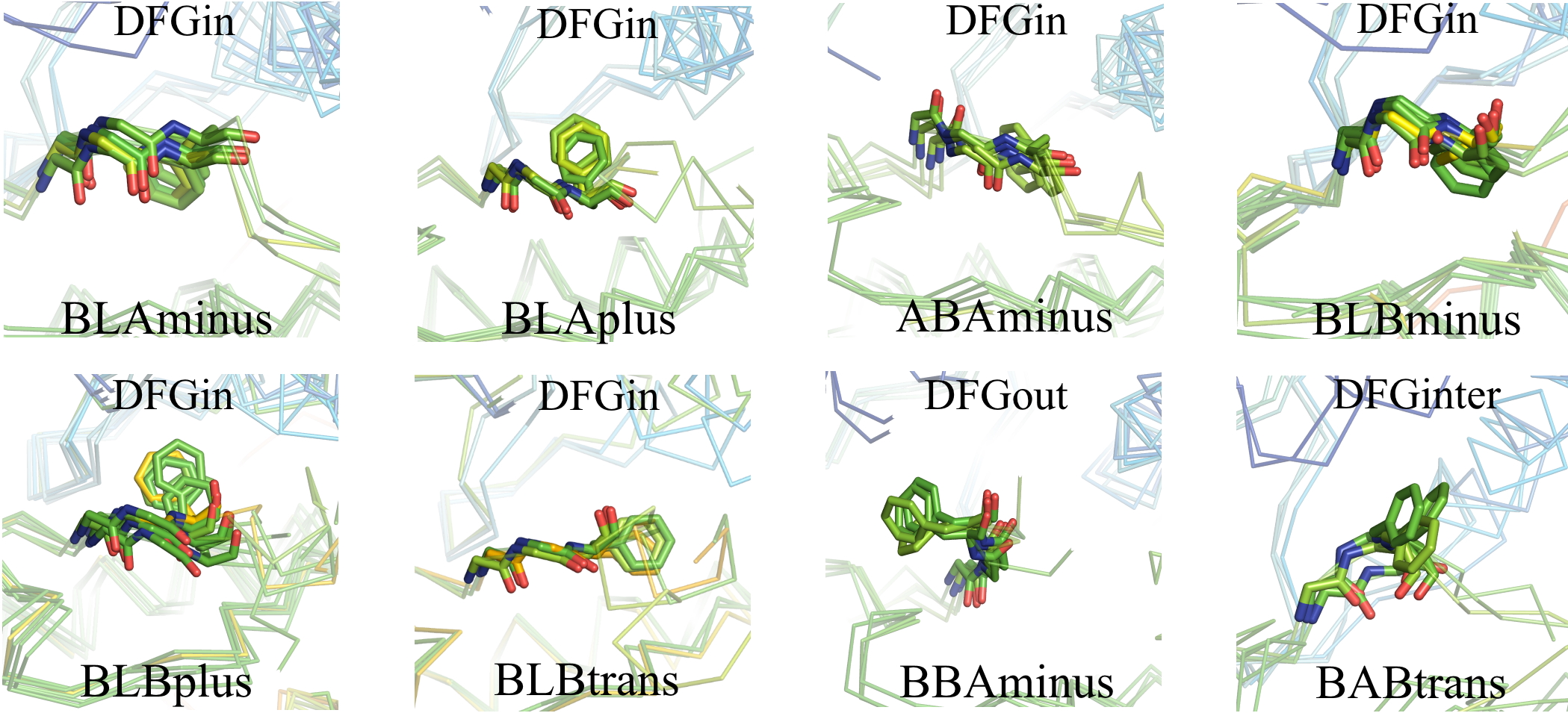
Dihedral clusters
Each spatial group consists of multiple closely related conformations. To cluster these conformations we used the backbone dihedrals (φ,ψ) of X-DFG (residue before conserved Asp), DFG-Asp, DFG-Phe and side chain dihedral (χ1) of DFG-Phe. These dihedrals we used to compute a distance matrix which is then provided as an input to DBSCAN (Density-based spatial clustering of applications with noise), a density-based clustering algorithm. The different clusters observed are labeled on the basis of Ramachandran region (A, B, L, and E) occupied by XDF residue backbone and the DFG-Phe χ1 rotamer (minus = -60°; plus = +60°; trans = 180°).
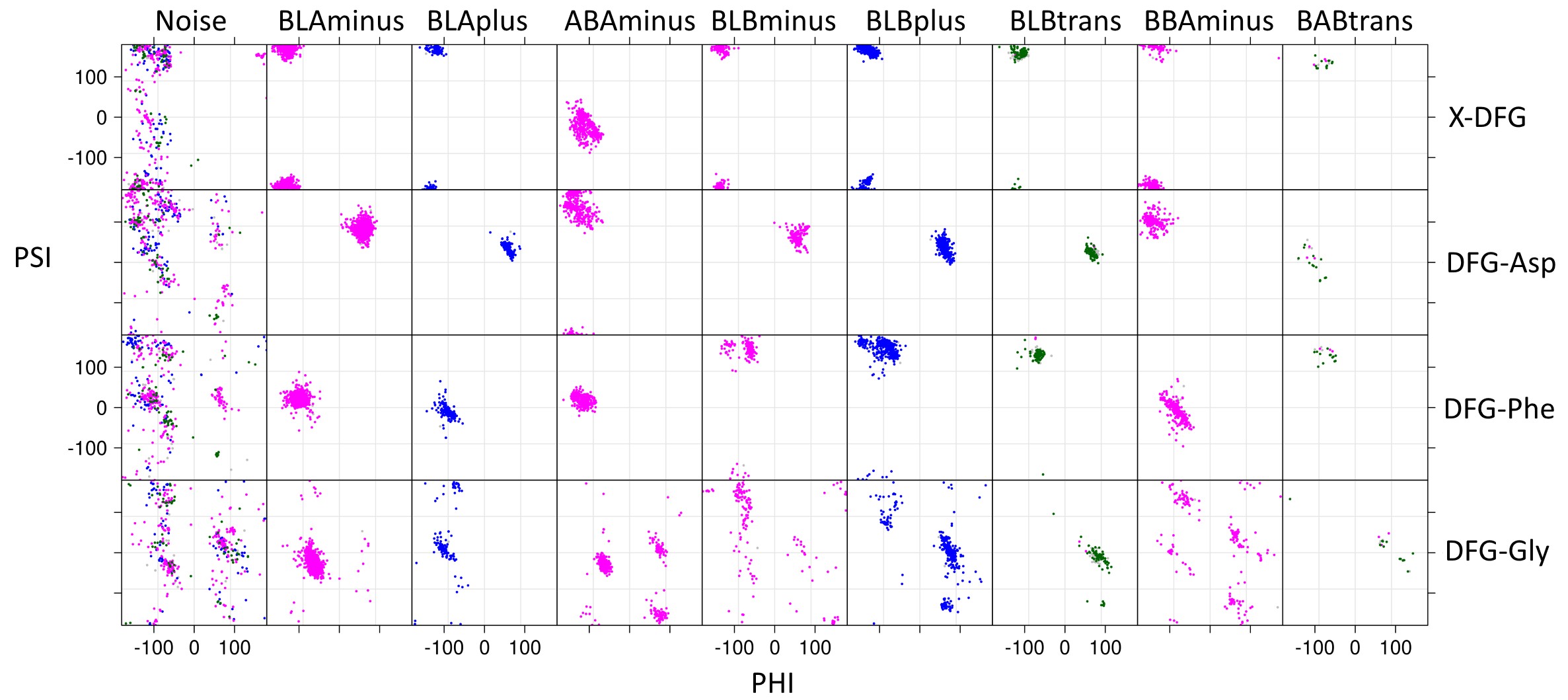
For the DFGin group we obtained six clusters labeled as BLAminus, BLAplus, ABAminus, BLBminus, BLBplus, and BLBtrans. All the catalytically primed structures (ATP+Mg bound and activation loop phosphorylated) are observed in the BLAminus cluster.
For the DFGout group we obtained just one cluster. In this cluster, the X-D-F residues occupy the B-B-A regions of the Ramachandran map and DFG-Phe is in a -60° rotamer. More than 82% of Type 2 inhibitor bound structures are BBAminus; the remainder are in the DFGout noise group.
The structures in the DFGinter conformation display more variability than the other states. For the DFGinter group we obtained only one small cluster. The X-D-F residues are in a B-A-B conformation and the DFG-Phe residue is observed in a trans rotamer with a few chains displaying a rotamer orientation between g-minus and trans.
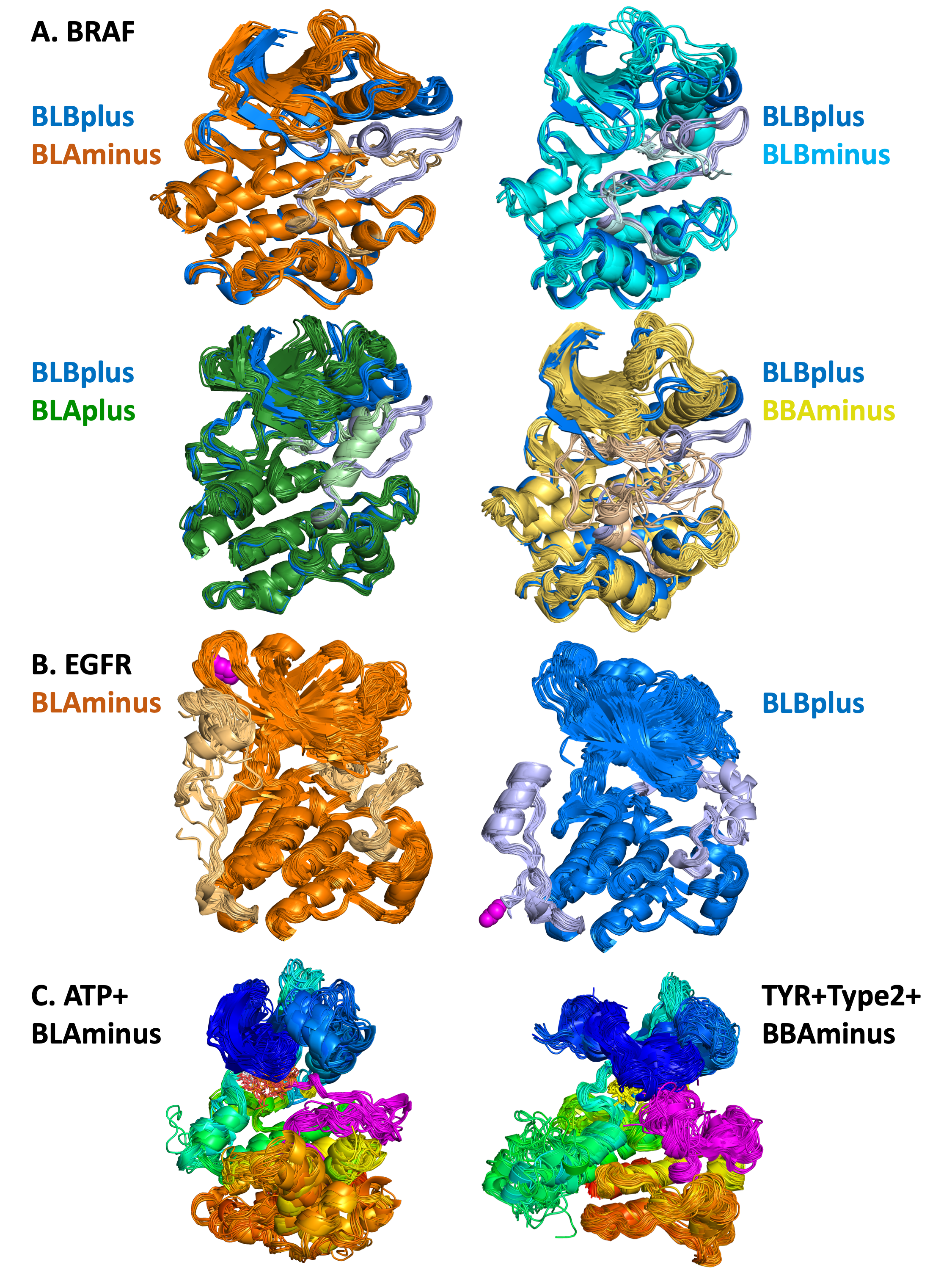
A. Pairwise comparisons of 5 different states of human BRAF kinase
DFGin-BLBplus structures (“SRC-inactive” conformation in blue with grayish-blue activation loop)
compared with DFGin-BLAminus (orange, active conformations), BLBminus (magenta-pink), DFGinBLAplus (“FGFR-inactive”, cyan-lightcyan), and DFGout-BBAminus (Type-2 ligand binding, lightbrownyellow). A small number of outlier structures in some classes are not shown. In each case, the position of the activation loop and the C-helix is correlated with
the state of the kinase, and differs among the 5 states.
B. BLAminus and BLBplus states of EGFR
EGFR BLAminus (107
chains, left, orange) and BLBplus (79 chains, right, blue) structures with both the activation loop
(right side of each image) and C-terminal tail (left side of each image) shown in lighter colors. The C-terminus of each group is shown in magenta spheres. The state of the activation loop is highly correlated with the
position of the C-terminal tail. In the active BLAminus structures, the tail is mostly coil and reaches up
to strands B2 and B3 of the N-terminal domain. In the “SRC-inactive” BLBplus state, the tail contains a
helix (residues 993-1002) in contact with the C-terminal domain, and then turns around with the C-terminus in contact with the I-helix of the C-terminal domain.
C. Results of searches with the Kincore website
ATP-bound BLAminus structures from 14
different kinases in 6 kinase families (left) and BBAminus structures from 29 kinases with bound
Type 2 inhibitors (right).In the
BLAminus structures, the position of the DFG Phe (in yellow) and the conformation of the DFGmotif at
the beginning of the activation loop (shown in magenta) and the overall position of the activation loop
are consistent across the structures.
Protein kinase inhibitors
We have classified PK inhibitors into five groups based on the region of protein they bind to.
- Type 1
- Type 1.5 - subdivided as Type 1.5_Front and Type 1.5_Back
- Type 2
- Type 3
- Allosteric
A list of FDA approved PK inhibitors with known structures can be accessed here.